

Trong sản xuất dược phẩm, đặc biệt đối với các sản phẩm vô trùng, endotoxin là một trong những nguồn nguy cơ nhiễm bẩn khó phát hiện nhất. Không màu, không mùi và không thể phát hiện bằng mắt thường và chúng không thể bị loại bỏ bởi các phương pháp khử khuẩn thông thường. Chỉ một sơ xuất nhỏ khi kiểm soát endotoxin trong sản xuất Dươc Phẩm là đủ để Độc tốnày xâm nhập vào sản phẩm tiêm truyền gây phản ứng sinh sốt (pyrogenic reactions) và trong các trường hợp nghiêm trọng có thể đe dọa tính mạng người bệnh.
Bài viết này giải thích endotoxin là gì, cách kiểm soát endotoxin trong sản xuất Dược Phẩm và môi trường phòng sạch dược phẩm, cũng như các dẫn chứng từ FDA.
Endotoxin là gì — Định nghĩa chính xác
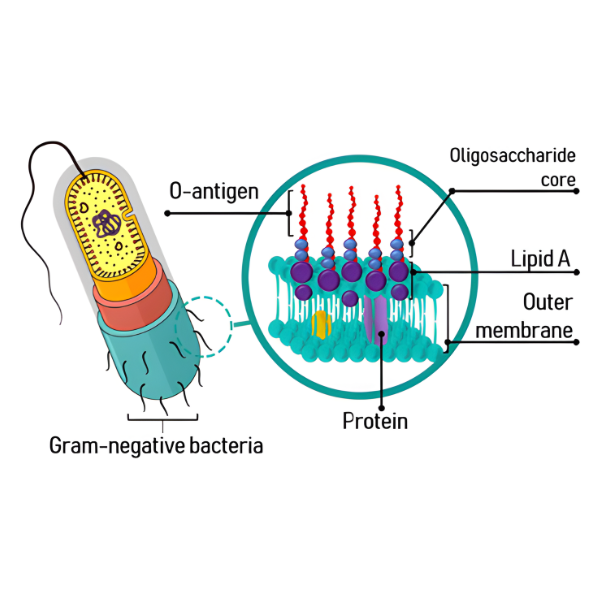
Source ảnh Cấu trúc của Endotoxin: https://www.mdpi.com/2072-6651/13/8/533
Endotoxin, hay nội độc tố của vi khuẩn Gram âm, là thành phần lipopolysaccharide (LPS) cấu tạo nên màng ngoài của vi khuẩn Gram âm như Escherichia coli, Pseudomonas aeruginosa và Salmonella spp. Endotoxin được giải phóng khi tế bào vi khuẩn chết, bị ly giải hoặc bị phân hủy
Endotoxin trở thành mối nguy đặc biệt trong sản xuất dược phẩm do bốn đặc tính quan trọng sau:
- Endotoxin có độ bền nhiệt cao và không bị phá hủy ở nhiệt độ khử trùng bằng hơi nước thông thường (121°C trong autoclave).
- Endotoxin không bị giữ lại bởi màng lọc vi khuẩn 0,22 µm, vì kích thước phân tử nhỏ hơn tế bào vi khuẩn.
- Endotoxin có thể tích lũy trong hệ thống nước dược dụng và trên bề mặt thiết bị nếu chương trình kiểm soát không được thực hiện đầy đủ.
- Khi xâm nhập vào cơ thể qua đường tiêm, endotoxin có thể gây phản ứng sinh sốt (pyrogenic reaction) ngay cả ở nồng độ rất thấp.
Phân biệt Endotoxin, Bioburden và Pyrogen

Bảng bên dưới sẽ tóm tắt 03 khái niệm trong thực hành sản xuất và kiểm nghiệm dược phẩm – mặc dù bản chất, phương pháp để phát hiện và xử lý hoàn toàn khác nhau:
| Tiêu chí | Endotoxin | Bioburden | Pyrogen |
| Bản chất | LPS từ màng vi khuẩn Gram âm | Tổng vi sinh vật sống trong mẫu | Bất kỳ chất nào gây sốt khi vào cơ thể |
| Phương pháp để nhận biết | BET / LAL test (USP <85>) | Đếm khuẩn lạc, lọc màng | Thử nghiệm, MAT |
| Bị loại bỏ bằng phương pháp khử khuẩn | Không | Có | Tùy loại |
| Lọc 0,22 µm giữ lại | Không | Có | Tùy loại |
| Mối quan hệ | Là một loại pyrogen | Có nguy cơ hình thành endotoxin khi tăng cao kéo dài | Khái niệm rộng hơn endotoxin |
Nguồn tham khảo: https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/inspection-technical-guides/bacterial-endotoxinspyrogens
Nguồn tham khảo: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/guidance-industry-pyrogen-and-endotoxins-testing-questions-and-answers
Điểm thực hành quan trọng: Kết quả bioburden đạt yêu cầu không đồng nghĩa với việc endotoxin cũng đạt yêu cầu. Endotoxin có thể tồn tại trong hệ thống ngay cả khi bioburden đã được kiểm soát, vì endotoxin không bị loại bỏ bởi các tác nhân khử khuẩn thông thường.
Xét nghiệm endotoxin là gì — BET và LAL test

Để phát hiện và định lượng endotoxin trong sản phẩm, các cơ sở sản xuất dược phẩm thường sử dụng phương pháp Thử nội độc tố vi khuẩn (Bacterial Endotoxins Test — BET), còn được gọi là LAL test (Limulus Amebocyte Lysate test).
Theo tài liệu thanh tra kỹ thuật của FDA:
“Bacterial Endotoxins Test, also termed the Limulus Amebocyte Lysate (LAL) Test.”
Link tham khảo: https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/inspection-technical-guides/bacterial-endotoxinspyrogens
Nguyên lý của LAL test dựa trên phản ứng đông đặc của dịch chiết từ tế bào amebocyte của cua móng ngựa (Limulus polyphemus) khi tiếp xúc với endotoxin. Đây là phương pháp nhạy, đặc hiệu và được chuẩn hóa trong USP <85> Bacterial Endotoxins Test.
FDA cũng ban hành hướng dẫn riêng cho ngành dược phẩm, sinh phẩm và thiết bị y tế liên quan đến việc kiểm nghiệm endotoxin và tiêu chí chấp nhận theo USP <85>.
Link tham khảo: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/guidance-industry-pyrogen-and-endotoxins-testing-questions-and-answers
Ba phương pháp BET phổ biến hiện nay:
| Phương pháp | Nguyên lý | Ứng dụng phổ biến |
| Gel-clot | Quan sát đông đặc gel | Định tính, bán định lượng |
| Turbidimetric | Đo sự thay đổi độ đục | Định lượng |
| Chromogenic | Đo màu sinh ra từ phản ứng enzyme | Định lượng độ nhạy cao |
Nguồn tham khảo: https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/inspection-technical-guides/bacterial-endotoxinspyrogens
Nguồn tham khảo: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/guidance-industry-pyrogen-and-endotoxins-testing-questions-and-answers
Endotoxin trong nước WFI và PW — Tại sao hệ thống nước là điểm kiểm soát trọng yếu
Hệ thống nước là một trong những nguồn phát sinh endotoxin phổ biến nhất trong nhà máy dược phẩm. Hai loại nước cần đặc biệt lưu ý gồm:
Nước tinh khiết (Purified Water — PW)
Purified Water không có giới hạn endotoxin compendial theo USP, vì loại nước này chủ yếu được sử dụng cho các ứng dụng không vô trùng. Tuy nhiên, nhiều nhà máy vẫn theo dõi endotoxin như một chỉ số kiểm soát nội bộ, đặc biệt trong các hệ thống nước có nguy cơ phát sinh vi khuẩn Gram âm. Nếu hệ thống PW bị ô nhiễm sinh học, endotoxin có thể tích lũy trong hệ thống phân phối nước, đường ống và các điểm lấy mẫu.
Nước pha tiêm (Water for Injection — WFI)
Theo USP và EP, giới hạn endotoxin của WFI là ≤ 0.25 EU/mL. Trong thực tế vận hành, nhiều nhà máy thiết lập giới hạn kiểm soát nội bộ thấp hơn để duy trì trạng thái kiểm soát của hệ thống nước. WFI được sử dụng trực tiếp trong pha chế thuốc tiêm và dung dịch truyền, vì vậy bất kỳ sự hiện diện của endotoxin trong công đoạn này đều có thể gây hậu quả nghiêm trọng cho người bệnh.
Để hiểu rõ hơn về cách WFI được kiểm soát toàn diện trong nhà máy dược GMP, bao gồm cả chiến lược xử lý OOS và OOT, bạn có thể tham khảo thêm tại: Nước WFI là gì và dùng thế nào trong nhà máy dược GMP
Nguồn gây nhiễm endotoxin trong sản xuất dược phẩm
Kiểm soát endotoxin hiệu quả bắt đầu từ việc xác định đúng các nguồn phát sinh endotoxin trong hệ thống sản xuất. Trong thực tế vận hành nhà máy dược, năm nguồn sau thường được xem là các điểm nguy cơ chính cần kiểm soát.
- Hệ thống nước dược dụng là một trong những nguồn phát sinh endotoxin quan trọng và khó kiểm soát nhất. Các điểm nước đọng, đoạn ống chết (dead leg) trong đường ống và vòng tuần hoàn không duy trì đủ vận tốc dòng chảy là nơi vi khuẩn Gram âm dễ tích tụ và phát triển.
- Nguyên liệu thô và tá dược cần được đánh giá và kiểm soát endotoxin trong quá trình tiếp nhận và thẩm định nguyên liệu, đặc biệt là các nguyên liệu có nguồn gốc sinh học hoặc tự nhiên vì đây là nguồn endotoxin nội sinh khó dự đoán.
- Thiết bị và bề mặt tiếp xúc sản phẩm là các điểm rủi ro cần được thẩm định riêng. Endotoxin có thể bám dính trên bề mặt inox, thủy tinh và polymer nếu quy trình làm sạch không đủ hiệu lực để loại bỏ lipopolysaccharide (LPS)
- Bao bì sơ cấp bao gồm lọ thủy tinh, túi IV và nút cao su cần được xử lý khử nội độc tố (depyrogenation) trước khi đưa vào dây chuyền sản xuất.
- Môi trường phòng sạch là nguồn phát sinh endotoxin gián tiếp thường bị đánh giá thấp trong chương trình kiểm soát vi sinh. Vi khuẩn Gram âm trong không khí hoặc do nhân sự mang vào có thể trở thành điểm khởi đầu của chuỗi nhiễm nếu chương trình giám sát vi sinh không đủ nhạy để phát hiện sớm.
Cách kiểm soát endotoxin trong hệ thống nước và nhà máy

Kiểm soát endotoxin không phải là một biện pháp đơn lẻ mà là một hệ thống phòng ngừa nhiều lớp trong toàn bộ quy trình sản xuất.
Với hệ thống nước, nguyên tắc vận hành cốt lõi là không để nước đứng yên. Vòng tuần hoàn liên tục với tốc độ dòng chảy đủ tiêu chuẩn là hàng rào đầu tiên chống lại sự hình thành màng vi sinh (biofilm) và tích lũy endotoxin. Khử trùng định kỳ bằng nhiệt hoặc hóa chất theo SOP được thẩm định là yêu cầu bắt buộc. Dữ liệu lấy mẫu tại các điểm sử dụng cần được phân tích xu hướng liên tục để phát hiện sớm dấu hiệu bất thường trước khi chạm ngưỡng tiêu chuẩn OOS.
Với thiết bị và bề mặt, quy trình vệ sinh phải được thẩm định để chứng minh hiệu lực loại bỏ endotoxin không chỉ loại bỏ vi sinh vật. Tiêu chí nghiệm thu phải bao gồm cả chỉ tiêu endotoxin khi áp dụng cho thiết bị tiếp xúc sản phẩm vô trùng. Về cách xây dựng SOP vệ sinh đúng chuẩn cGMP, thiết lập tiêu chí nghiệm thu và đo lường KPI vệ sinh, bạn có thể đọc thêm tại: Quy Trình Vệ Sinh Nhà Máy Theo GMP EU
Với môi trường phòng sạch, chương trình giám sát vi sinh môi trường cần được thiết kế để phát hiện sự gia tăng của vi khuẩn Gram âm, vì đây là chỉ báo gián tiếp quan trọng về nguy cơ nhiễm endotoxin. Có thể tham khảo thêm cách triển khai chương trình kiểm soát vi sinh toàn diện tại: Kiểm soát vi sinh tại phòng sạch
Endotoxin OOS là gì và xử lý thế nào
Kết quả OOS (Out of Specification) về endotoxin là tình huống kết quả kiểm nghiệm vượt giới hạn cho phép đã được thiết lập trong hồ sơ sản phẩm hoặc tiêu chuẩn dược điển.
Đây là tình huống phải được điều tra theo quy trình OOS chính thức. Các dẫn chứng từ FDA cho thấy hậu quả nghiêm trọng khi kết quả endotoxin OOS không được xử lý đúng cách:
“Released the above batches with OOS endotoxin failures for U.S. distribution.”
“They should not be the sole basis for batch release.”
— FDA Warning Letter, Liebel-Flarsheim Company LLC (10/17/2025)
Trường hợp này cho thấy việc phát hành lô sản phẩm khi có kết quả endotoxin OOS là vi phạm nghiêm trọng nguyên tắc kiểm soát chất lượng. Đối với sản phẩm tiêm truyền, điều này có thể dẫn đến nguy cơ phản ứng sinh sốt (pyrogenic reaction) ở bệnh nhân, đồng thời khiến doanh nghiệp đối mặt với các biện pháp quản lý như thu hồi sản phẩm, cảnh báo nhập khẩu hoặc đình chỉ sản xuất.
Link tham khảo: https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/warning-letters/liebel-flarsheim-company-llc-711508-10172025
Quy trình xử lý endotoxin OOS theo cGMP thường bao gồm 4 bước chính.
- Bước đầu tiên là dừng lô và không xuất xưởng trong khi đang điều tra.
- Bước thứ hai là điều tra tại phòng kiểm nghiệm để xác minh lỗi có phải do quá trình kiểm nghiệm không, bao gồm mẫu, thuốc thử, thiết bị và kỹ thuật viên.
- Bước thứ ba là điều tra mở rộng toàn diện nếu đã loại trừ lỗi tại phòng kiểm nghiệm, mở rộng phạm vi điều tra ra quy trình sản xuất, hệ thống nước, vệ sinh thiết bị và nguyên liệu.
- Bước cuối cùng là CAPA và đánh giá hiệu lực, xác định nguyên nhân gốc rễ thực sự, triển khai hành động khắc phục và xác nhận lỗi không tái phát.
Một sai lầm phổ biến là mở điều tra OOS chỉ để hoàn tất hồ sơ mà không xác định được nguyên nhân phát nguồn. FDA đánh giá rất nặng điều này, như đã ghi nhận trong thư cảnh báo gửi đến Advanced Pharmaceutical Technology (03/2025), khi FDA nhận xét phản hồi của doanh nghiệp về kế hoạch kiểm soát tạp nhiễm vi sinh và endotoxin là chưa đủ chi tiết và chưa giải quyết được gốc rễ vấn đề:
“Plan to test for bioburden, prior to release, and will reject lots… for endotoxin.”
— FDA Warning Letter, Advanced Pharmaceutical Technology (03/14/2025)
Cách xử lý tín hiệu Endotoxin theo tình huống

Thay vì chỉ dò theo các checklist cứng nhắc, đội QA, QC và đội Kỹ thuật nên áp dụng ma trận triệu chứng – giả thuyết – hành động để làm việc trên cùng một hệ quy chiếu. Cách tiếp cận này buộc đội ngũ phải đặt câu hỏi đúng trước khi hành động, thay vì phản ứng theo cảm tính hoặc theo thói quen.
Tình huống 1: Endotoxin tăng nhẹ, chưa OOS, có dấu hiệu lệch xu hướng
Giả thuyết hay gặp nhất là thao tác lấy mẫu thiếu ổn định, điểm lấy mẫu không mang tính đại diện tổng quan, hoặc hệ thống đang xuống cấp sau các đợt bảo trì gần nhất. Hành động cần thực hiện là khóa chặt điều kiện lấy mẫu và xét nghiệm, tiến hành lấy mẫu lại có kiểm soát, rà soát xu hướng theo từng điểm lấy riêng biệt và đối chiếu với các sự kiện vận hành nhà máy trong cùng thời kỳ.
Tình huống 2: Endotoxin OOS đơn lẻ, các chỉ tiêu khác chưa báo động
Giả thuyết hay gặp nhất là sai lệch không ổn định trong quá trình xét nghiệm mẫu thử, nhân viên pha loãng hoặc xử lý mẫu sai nhánh SOP, hoặc vô tình gây nhiễm chéo trong lúc thao tác. Hành động đúng là giữ nguyên mẫu thử sai lệch, rà soát toàn bộ điều kiện thực hiện xét nghiệm và xác minh tất cả các khả năng có thể gây sai lệch theo yêu cầu của phương pháp trước khi quyết định mở rộng phạm vi điều tra ra bên ngoài phòng kiểm nghiệm.
Tình huống 3: Endotoxin OOS kèm tín hiệu tạp nhiễm vi sinh hoặc môi trường xấu đi
Đây là tình huống nghiêm trọng nhất. Giả thuyết hay gặp là hệ thống kiểm soát tạp nhiễm đang thất bại, đường ống nước hoặc thiết bị có điểm tạo điều kiện cho vi sinh vật phát triển, hoặc màng vi sinh (biofilm) đang bùng phát. Hành động phải khởi động ngay quy trình xử lý sai lệch, khoanh vùng các điểm áp dụng quy trình, tăng tần suất lấy mẫu giám sát, đánh giá tác động lên các lô sản phẩm liên quan và đồng thời rà soát lại toàn diện chiến lược khử trùng hiện tại.
Tình huống 4: Endotoxin tăng sau bảo trì, thay vật liệu hoặc thay cấu hình đường ống
Giả thuyết hay gặp là cặn bẩn và lớp bám lâu ngày bị bong tróc ra, làm khuấy động quy trình tráng rửa sau bảo trì. Hành động cần áp dụng quy trình xử lý theo sự kiện, tăng cường các bước tráng rửa và khử trùng chuẩn SOP, đồng thời theo dõi dữ liệu sát sao hơn trong suốt giai đoạn phục hồi cho đến khi xu hướng ổn định trở lại.
Chốt ghi nhớ: Điều tra endotoxin OOS là một quy trình xử lý lớn được áp dụng vào hệ thống kiểm soát. Đây không phải là hành trình đi tìm một cái cớ để hợp thức hóa cho một kết quả đạt.
FAQ về Endotoxin trong sản xuất Dược Phẩm
Endotoxin là gì?
Endotoxin hay nội độc tố vi khuẩn Gram âm là thành phần lipopolysaccharide (LPS) trong màng ngoài của vi khuẩn Gram âm, được giải phóng khi vi khuẩn chết. Endotoxin gây phản ứng sốt nguy hiểm khi vào cơ thể qua đường tiêm.
Xét nghiệm endotoxin là gì?
Xét nghiệm endotoxin hay BET (Bacterial Endotoxins Test) là phương pháp kiểm nghiệm sử dụng dịch chiết LAL để phát hiện và định lượng endotoxin trong sản phẩm dược, nước và nguyên liệu, theo tiêu chuẩn USP <85>.
Giới hạn endotoxin trong nước WFI và PW là bao nhiêu?
Theo USP, nước PW có giới hạn endotoxin ≤ 0,25 EU/mL. Nước WFI có giới hạn < 0,25 EU/mL và trong thực tế vận hành thường được kiểm soát chặt hơn giới hạn pháp lý.
Endotoxin OOS phải xử lý thế nào?
Cần dừng lô, tiến hành điều tra 2 giai đoạn gồm điều tra tại phòng kiểm nghiệm và điều tra mở rộng toàn diện, xác định nguyên nhân gốc rễ thực sự, triển khai CAPA và đánh giá hiệu lực sau một thời gian áp dụng. Tuyệt đối không xuất xưởng lô khi chưa có kết luận điều tra đầy đủ.
Endotoxin khác gì Pyrogen và Bioburden?
Endotoxin là một loại pyrogen có nguồn gốc từ vi khuẩn Gram âm. Pyrogen là khái niệm rộng hơn, bao gồm mọi chất gây sốt khi vào cơ thể. Bioburden là tổng lượng vi sinh vật sống trong mẫu và là chỉ báo gián tiếp của nguy cơ endotoxin mức độ tạp nhiễm vi sinh tăng cao kéo dài mà không kiểm soát sẽ dẫn đến tích lũy endotoxin, nhưng tạp nhiễm vi sinh đạt chuẩn không đảm bảo endotoxin đạt chuẩn.
Nguồn gây nhiễm endotoxin trong nhà máy dược là gì?
Năm nguồn chính gồm hệ thống nước, nguyên liệu thô và tá dược, thiết bị và bề mặt tiếp xúc sản phẩm, bao bì sơ cấp và môi trường phòng sạch. Trong đó hệ thống nước là nguồn lây nhiễm phổ biến và khó kiểm soát nhất vì liên quan trực tiếp đến cấu hình đường ống và chế độ vận hành liên tục.
Kết luận
Endotoxin là mối nguy tiềm ẩn đặc thù của ngành dược không nhìn thấy, không ngửi được, nhưng hậu quả để lại với người bệnh là không thể đảo ngược. Kiểm soát endotoxin hiệu quả đòi hỏi sự phối hợp đồng bộ giữa hệ thống nước được vận hành đúng chuẩn, quy trình vệ sinh có hiệu lực được thẩm định, chương trình giám sát vi sinh chủ động và cơ chế xử lý OOS đủ chiều sâu để tìm ra nguyên nhân gốc rễ thực sự.
Bài học từ các dẫn chứng từ FDA rất rõ ràng: xuất xưởng lô khi có kết quả endotoxin OOS hoặc xử lý OOS không đủ căn cứ khoa học là vi phạm cGMP nghiêm trọng và FDA không bỏ qua điều đó.



